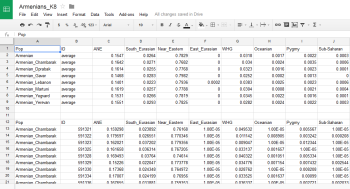
The Armenian samples from the recent Haber et al. manuscript on the genetic history of Armenia are now freely available at the Wellcome Trust Sanger website here.
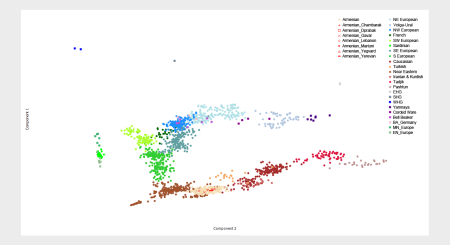
I ran an analysis of 127 of these Armenians using my K8 model, after removing a couple of the individuals from Chambarak with obvious European admixture, as well as a few other problem samples. The results are shown below, along with maps of the sampling locations (courtesy of Wikipedia).
It seems that, at least based on this dataset, Armenians are a fairly homogenous bunch of people. The two groups from east of Lake Sevan show slightly higher levels of ANE than most of the rest, but this is in line with geography, because they're located a little closer to the Northeast Caucasus, where ANE peaks in West Eurasia at almost 30%.
By the way, those of you having trouble with the acronyms in this and other recent blog posts, you'll find a list of often used Eurogenes acronyms here.
See also...
The Near East ain't what it used to be
I have a feeling that this generally underrated phenomenon will become much less underrated in the near future, and might even be recognized as a major factor in the formation of the modern European gene pool.
Ancient human mobility at the individual level is conventionally studied by the diverse application of suitable techniques (e.g. aDNA, radiogenic strontium isotopes, as well as oxygen and lead isotopes) to either hard and/or soft tissues. However, the limited preservation of coexisting hard and soft human tissues hampers the possibilities of investigating high-resolution diachronic mobility periods in the life of a single individual. Here, we present the results of a multidisciplinary study of an exceptionally well preserved circa 3.400-year old Danish Bronze Age female find, known as the Egtved Girl. We applied biomolecular, biochemical and geochemical analyses to reconstruct her mobility and diet. We demonstrate that she originated from a place outside present day Denmark (the island of Bornholm excluded), and that she travelled back and forth over large distances during the final months of her life, while consuming a terrestrial diet with intervals of reduced protein intake. We also provide evidence that all her garments were made of non-locally produced wool. Our study advocates the huge potential of combining biomolecular and biogeochemical provenance tracer analyses to hard and soft tissues of a single ancient individual for the reconstruction of highresolution human mobility.
Frei, K.M. et al. Tracing the dynamic life story of a Bronze Age Female. Sci. Rep. 5, 10431; doi: 10.1038/srep10431 (2015).
At the transition from the third to the second millennium BC, the introduction of bronze for the manufacture of tools, weapons and personal ornaments marked a major step in European prehistory. Trade of the metal raw materials and manufactured goods required regular and organized contacts among communities. On the other hand, local population continuity was a prerequisite for the accumulation of wealth, the establishment of enduring social differentiation, and the formation of regional elites.
The archeological record in the Lech Valley in southern Bavaria, Germany, shows a rapid and gapless transition from the Late Neolithic Bell Beaker Phenomenon to the Early Bronze Age. To investigate social and demographic changes associated with the appropriation of the bronze technology, we studied nearly eighty individuals from six burial sites in the region with respect to their maternally inherited mitochondrial DNA (mtDNA).
The results indicate both local genetic continuity spanning the cultural transition, and, following the onset of the Early Bronze Age, a major influx of mtDNA types previously not found in this region. Integrating stable isotope data with the genetic data reveals a picture of a patrilocal society with remarkable mobility in women. While crucial for understanding the change of local demographics, these findings also have implications for the spread of major technological and societal changes across Europe at the beginning of the Bronze Age.
Mittnik et al., Ancient DNA reveals patterns of residential continuity and mobility at the onset of the Central European Bronze Age, Society for Molecular Biology and Evolution (SMBE) 2015 abstract
See also...
Female mobility and exogamy as the main drivers of foreign admixture during the Late Neolithic/Early Bronze Age shift in Central Europe (Knipper et al. 2017)
Corded Ware women more mobile than their men (Sjögren et al. 2016)
Lots of paleogenomics stuff this year. Good to see. The abstract book is available here. Below are a few highlights:
Genomic signals of migration and continuity in Roman Britain
Martiniano et al.
York (Eboracum) was a provincial city at the edge of the Roman Empire where a number of Roman cemeteries have been excavated. One of these at Driffield Terrace is unusual in a regional and national context, with a large predominance (70.8%) of decapitated young males buried there. They show frequent evidence for trauma consistent with violent life histories and have been alternately speculated as gladiators, soldiers and slaves with potentially foreign origins. Here we report the ~1X genome sequences of seven of these individuals from the Roman period. While six of the Roman burials show affinity with modern British populations, one sample, although indistinguishable in funerary ritual from the other skeletons in the cemetery, shows a clear signal of exogenous origin, with modern affinities pointing towards the Eastern Mediterranean, a clear indication of the cosmopolitan impetus of the Roman empire, even at its western fringe.
Insights into British and European population history from ancient DNA sequencing of Iron Age and Anglo-Saxon samples from East England
Schiffels et al.
British population history is shaped by a series of immigration periods and associated changes in population structure. It is an open question to what extent these changes affect the genetic composition of the current British population. Here we present whole genome sequences generated from 10 individuals, found in archaeological excavations in Hinxton, Oakington and Linton, close to Cambridge, and ranging from 2,300 years before present (Iron Age) until 1,200 years before present (Anglo-Saxon period). We use modern genetic samples from the 1000 Genomes Project and additional external data from Britain, the Netherlands and Denmark to characterize the relationship of these ancient samples with contemporary British and other European populations. By analyzing the distribution of shared rare variants across ancient and modern individuals, we find that samples from the Anglo-Saxon period are relatively more closely related to central northern Europe, while earlier samples and contemporary British samples are relatively more closely related to Southern European populations. To quantify this series of relationships further, we developed a new method, rarecoal, that fits a demographic model parameterized by split times, population sizes and migration rates to the distribution of shared rare variants across a large number of modern and ancient individuals. We use rarecoal to estimate the history of European population structure within the last 10,000 years and to map our ancient samples onto the European population tree. Our approach provides a unique picture of population history in Europe, and in particular helps characterizing the complex genetic impact of Anglo-Saxon immigrations into Britain.
Ancient DNA reveals patterns of residential continuity and mobility at the onset of the Central European Bronze Age
Mittnik et al.
At the transition from the third to the second millennium BC, the introduction of bronze for the manufacture of tools, weapons and personal ornaments marked a major step in European prehistory. Trade of the metal raw materials and manufactured goods required regular and organized contacts among communities. On the other hand, local population continuity was a prerequisite for the accumulation of wealth, the establishment of enduring social differentiation, and the formation of regional elites.
The archeological record in the Lech Valley in southern Bavaria, Germany, shows a rapid and gapless transition from the Late Neolithic Bell Beaker Phenomenon to the Early Bronze Age. To investigate social and demographic changes associated with the appropriation of the bronze technology, we studied nearly eighty individuals from six burial sites in the region with respect to their maternally inherited mitochondrial DNA (mtDNA).
The results indicate both local genetic continuity spanning the cultural transition, and, following the onset of the Early Bronze Age, a major influx of mtDNA types previously not found in this region. Integrating stable isotope data with the genetic data reveals a picture of a patrilocal society with remarkable mobility in women. While crucial for understanding the change of local demographics, these findings also have implications for the spread of major technological and societal changes across Europe at the beginning of the Bronze Age.
The Genomic connections between early farmers from Iberia and central Europe, and their relationship to populations from modern Spain
Gunther et al.
The neolithization process swept over Europe after the advent of farming lifestyle in the Near East approximately 11,000 years ago. However, the mode of transition and its impact on the demographic patterns of Europe remains an area of open questions. Ancient genomics allow us to analyze individuals involved in these transitions directly and to make comparisons between populations over time. Previous studies have shown close relationships between early Scandinavian farmers and contemporary southern Europeans as well as strong differences between hunter gatherers and early farmers. However, mtDNA composition on the Iberian peninsula and different migration routes suggest a dissimilar history of southwestern Europe. We [obtained?] genomic sequences of eight between 4,000 and 5,600 year old early Iberian farmers from El Portalon, 15 km East of Burgos, Spain. In contrast to a 7,000 year old hunter gatherer from the near-by area La Brana, but similar to the pattern observed for central and northern European farmers, these individuals all show genetic similarities to modern-day southern Europeans, especially to Sardinians and Basques.
450 diverse high coverage whole genome sequences reveal ancient population admixture in modern human populations
Luca Pagani
Complete high-coverage individual genome sequences carry the maximum amount of information for reconstructing the evolutionary past of a species in the interplay between random genetic drift and natural selection. Here we present a novel dataset of 450 human genomes from 156 populations that represent a dense geographic coverage of Eurasia. The genomes, chosen to be representative of each population based on SNP-chip data, were sequenced at 40X on the same platform (Complete Genomics) and processed on a uniform bioinformatics pipeline.
Our dataset has an unprecedented combination of high spatial and genomic coverage. This enabled us to refine current knowledge on continent-wide patterns of heterozygosity, long and short range gene flow, archaic admixture, and changes in effective population size over time. In particular, we have clarified the admixture dynamics of Eurasian populations during the last 3000 years, confirmed and further resolved the genetic relationship between recently sequenced ancient human remains and modern populations, and highlighted significantly higher amounts of Neanderthal gene flows in Island South East Asian and Oceanian populations. We have also assembled an extensive catalogue of genes under positive selection in various human groups.
Finally, ChromoPainter (Lawson et al. 2012) and MSMC (Schiffels and Durbin 2014) have cemented genetic evidence of an early African origin for the people currently inhabiting Papua New Guinea. Our results are compatible with a first migration out of Africa of these Oceanian populations, which subsequently experienced 80% of gene flow from populations coming from the second, main Eurasian out of Africa.
Nuclear and mitochondrial DNA sequences from two Denisovan individuals
Sawyer et al.
Denisovans are a sister-group of Neandertals that were described based on a nuclear genome sequence from a finger phalanx (Denisova 3) found in Denisova Cave, Altai Mountains. A molar (Denisova 4) found at the same site, has a mitochondrial (mt) DNA sequence similar to Denisova 3. Here we present nuclear DNA sequences from Denisova 4, with the morphological description and the mitochondrial and nuclear DNA sequences from another molar (Denisova 8) from Denisova Cave. Like Denisova 4, this molar is very large and lacks traits typical of Neandertals and modern humans. Nuclear DNA sequences from the two molars form a clade with Denisova 3. The nuclear DNA sequence diversity among the three Denisovans is comparable to that among six Neandertals but lower than that among present-day humans. The mtDNA of Denisova 8 is more diverged from and has accumulated fewer substitutions than the mtDNAs of the other two specimens suggesting that Denisovans were present in the cave over an extended period of time.
Open access at Nature Communications today: Large-scale recent expansion of European patrilineages shown by population resequencing by Batini et al.
It's a shame the authors failed to sample any Eastern European populations, but it's still a very useful effort which moves us in the right direction in an area of study that has floundered from the start, largely due to the widespread use of bikini STR haplotypes and faulty methodology. From the paper:
Here, we use targeted NGS of European and Middle Eastern populations to show that Europe was affected by a major continent-wide expansion in patrilineages that post-dates the Neolithic transition. Resequencing at high coverage of 3.7 Mb of MSY DNA, in each of 334 males comprising 17 population samples, defines an unbiased phylogeny containing 5,996 high-confidence single-nucleotide polymorphisms (SNPs). Dating indicates that three major lineages (I1, R1a and R1b), accounting for 64% of the sampled chromosomes, have very recent coalescent times, ranging between 3.5 and 7.3 KYA. In demographic reconstructions (17) a continuous swathe of 13/17 populations from the Balkans to the British and Irish Isles share similar histories featuring a minimum effective population size ~2.1–4.2 KYA, followed by expansion to the present. Together with other data on maternally inherited mtDNA (16, 18) and autosomal DNA (19), our results indicate a recent widespread male-specific phenomenon that may point to social selection, and refocuses interest on the social and population structure of Bronze Age Europe.
...
The shapes of different clades within the tree (Fig. 1a) vary greatly. Haplogroups E1b-M35, G2a-L31, I2-P215, J2-M172, L-M11 and T-M70 contain long branches with deep-rooting nodes, whereas I1-M253, N1c-M178, R1a-M198 and R1b-M269 show much shallower genealogies.
The recent and rapid continent-wide demographic changes we observe suggest a remarkably widespread transition affecting paternal lineages. This picture is confirmed in an independent analysis of MSY diversity in the pooled HGDP CEPH panel European samples (16), and is compatible with current (n=98) ancient DNA data for MSY (Fig. 3; Supplementary Table 8), in which hgs R1a, R1b and I1 are absent or rare in sites dating before 5 KYA, whereas hgs G2a and I2 are prevalent.
The period 4–5 KYA (the Early Bronze Age) is characterized by rapid and widespread change, involving changes in burial practices that might signify an emphasis on individuals or kin groups, the spread of horse riding, and the emergence of elites and developments in weaponry (35). In principle male-driven social selection (36) associated with these changes could have led to rapid local increases in the frequencies of introgressing haplogroups (34), and subsequent spread, as has been suggested for Asia (37). However, cultures across Europe remain diverse during this period; clarifying the temporal and geographical pattern of the shift will rely heavily on additional ancient DNA data.
Batini, C. et al. Large-scale recent expansion of European patrilineages shown by population resequencing. Nat. Commun. 6:7152 doi: 10.1038/ncomms8152 (2015).
See also...
R1a1a from an Early Bronze Age warrior grave in Poland
Massive migration from the steppe is a source for Indo-European languages in Europe (Haak et al. 2015 preprint)
It looks like we're about to see yet another paper on the origins of Ashkenazi Jews. A poster on the topic was presented this week at the Biology of Genomes conference, and is available for download here.
It's a very reasonable effort, perhaps the best one so far. However, I'm of the opinion that the genetic structure of the Near East has changed significantly since the Neolithic. If that's correct, then using modern samples from the Near East to estimate Near Eastern ancestry in Ashkenazi Jews might not work too well.
For instance, let's assume, just for the sake of argument, that ~2,000 years ago the Levant was home to a population genetically almost indistinguishable from present-day Cretans. This might mean that Ashkenazi Jews are much less than 50% European. But we won't know until we see some ancient DNA from the Near East, including from the remains of early Jews.
Citation...
James Xue, Itsik Pe’er, and Shai Carmi, The time and place of European gene flow into Ashkenazi Jews, Biology of Genomes 2015 poster presentation.